|
|
|
|
|
Case #1
|
Typical Clinical Presentation
A 17-year-old male presented with increasing pain in the left upper arm of approximately 3 months' duration
and a recent onset of low-grade fever. On physical examination, there was some local tenderness
and soft tissue swelling over the proximal and mid thirds of the left humerus.
- Most important here is the patient's age and short duration of symptoms. Note that clinical picture overlaps significantly with that of osteomyelitis.
|
|
Characteristic Radiological Findings
|
|
|
|
-
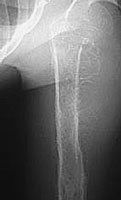
Plain radiograph shows a large ill-defined, destructive, diaphyseal intramedullary lesion
with permeative pattern of bone destruction and periosteal reaction of a "hair-on-end" type.
The lesion is associated with a soft tissue mass.
|
|
|
|
-
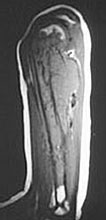
MRI is superior to the plain film in demostration of cortical disruption and soft tissue
involvement.
|
|
The major clue here is the intramedullary, diaphyseal location of the tumor. Although the
radiological features listed here (poor margination, permeative bone destruction, periosteal
"hair-on-end" reaction and soft tissue involvement) are very common in this entity, they are not entirely specific,
and just indicate the presence of a rapidly growing, most likely malignant, destructive tumor.
|
|
Pathological Findings
|
|
|
|
-
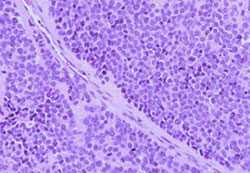
Biopsy material showed a highly cellular, infiltrative neoplasm consisting of sheets of
tightly packed, round cells with very scant cytoplasm ("round blue cell tumor").
Occasional Homer-Wright rosettes were identified. Other fields showed extensive necrosis.
|
|
|
|
-
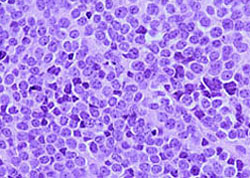
The cell population consisted of two distinct cell types:
the larger round cells with a high N/C ratio, fine chromatin pattern and occasional small,
inconspicuous nucleoli, and the smaller and darker cells with eosinophilic cytoplasm and
hyperchromatic, "shrunken" nuclei (degenerated cells, a typical finding in this entity).
Mitotic rate averaged 2 per 10 hpf.
|
|
|
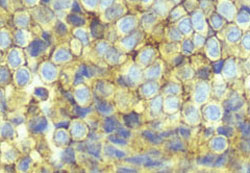
|
-
Tumor cells showed strong immunoreactivity with CD99/013. Neural markers (S-100; chromogranin
and synaptophysin) were uniformly negative.
|
|
|
|
The clue here is in the cytological appearance and pattern consisting
of sheets of primitive cells with little histologic evidence of differentiation.
Diagnosis of PNET as opposed to ES requires demonstration of neural differentiation,
which is evidenced histologically by formation of Homer-Wright rosettes (more than 20%)
and immunohistochemically by the expression of neural markers.
|
|
|
Diagnosis: Ewing's Sarcoma (ES)
|
|
Salient Points (Ewing's sarcoma and PNET)
|
- ES and PNET are "small round blue cell" tumors of children and young adults occurring in 80% of cases
between the ages of 5 and 20 years. They are extremely rare in patients older than 30 years. The difference between
the two is in the degree of neural differentiation. Some authors consider ES a tumor of undifferentiated neural cell,
which is of more primitive origin than cells of PNET or neuroblastoma.
- Most common skeletal sites include diaphyses of femur, tibia and humerus, and also pelvis and ribs
(Askin tumor of the chest). Associated soft tissue mass is a common finding.
- The following studies are required to support the diagnosis of ES and PNET:
Demonstration of t(11;22) or EWS-FLI-1 fusion transcript (present in both ES and PNET)
Immunostains(both ES and PNET are positive for CD99/O13. In addition, PNET shows positive staining
with neural markers)
EM (ES cells are undifferentiated and show prominent glycogen deposits; PNET shows neural differentiation)
- Aside from PNET, ES must be differentiated from other "small round blue cell" tumors:
|
|
Tumor
|
Positive immunostaing
|
|
ES
|
CD99/O13; may show positivity with NSE
|
|
PNET
|
CD99/O13
neurofilament; S-100; NSE; synaptophysin; chromogranin
|
|
Neuroblastoma
|
neurofilament; NSE; synaptophysin; chromogranin; S-100
|
|
Rhabdomyosarcoma
|
actin; desmin; vimentin; MyoD1; myogenin
|
|
Non-Hodgkin's lymphoma
|
CD45 (LCA)
|
|
Small cell carcinoma
|
keratin; NSE; synaptophysin; chromogranin
|
|
|
-
Studies have shown that occasionally ES/PNET may show strong immunoreactivity with cytokeratin
(AE1/AE3, Cam5.2) with either diffuse or focal staining pattern. Beware.
-
Prognostic factors. Disease stage at diagnosis (including the tumor volume) is the main prognostic factor for patients with ES/PNET; metastases are detected in about 15-20% of patients and portend a poor prognosis. Response to pre-operative chem
otherapy, as assessed by the degree of histologic tumor necrosis is a major independent prognostic factor. Histologic response to chemotherapy can be graded as follows: Grade I - tumor necrosis of less than 50%, Grade II - tumor necrosis of 51%-90, Gr
ade III - 91%-99% tumor necrosis, and Grade IV - absence of viable tumor cells (see reference below).
- Experimental data: Molecular prognostic factors for ES/PNET are being described. The most common and characteristic primary cytogenetic alteration seen in 95% of ES/PNET is t(11;22), which results in formation of the
chimeric gene/protein EWS-FLI-1, an abnormal transcription factor. About 5% of ES/PNET show t(21;22) with formation of analogous EWS-ERG fusion gene/protein. These EWS fusions are presumed to be the initiating oncogenic events in ES/PNET. The structure o
f fusion may have an independent prognostic significance. Recent studies have shown that a specific type of EWS-FLI-1 (type 1) is associated with better prognosis. Studies have also shown that during tumor progression, secondary molecular alterations may
occur, which often involve genes regulating cell cycle. Recent papers suggest that INK4A deletion (INK4A gene encodes a tumor supressor p16INK4A ) is a frequent secondary molecular alteration in ES/PNET which may be prognostically significant.
Aberrant p53 expression has been found in a small subset of patients with ES/PNET with a markedly poor clinical outcome.
|
|
|
|
Available publications for the topic:
Ewing's Sarcoma
Selected References::
|
- Wunder JS, Paulian G, Huvos AG, Heller G, Meyers PA, Healey J. The histological response to chemotherapy as a predictor of the oncological outcome of operative treatment of Ewing sarcoma. J Bone Joint Surg Am 1998;80A:1020-1033
- DeAlava E, Kawai A, Healey JH, Fligman I, Meyers P, Huvos AG, et al. EWS-FLI-1 fusion transcript structure is an independent determinant of prognosis in Ewing's sarcoma. J Clin Oncol 1998;16:1248-1255
- Alava E, Antonescu CR, Panizo A, Leung D, Meyers PA, Huvos AG, et al. Prognostic significance of p53 status in Ewing sarcoma. Cancer 2000;89(4):783-792
- Wei G, Antonescu CR, Alava E, Leung D, Huvos AG, Meyers PA, et al. Prognostic impact of INK4A deletion in Ewing sarcoma. Cancer 2000;89(4):793-799
- Paulussen M, Ahrens S, Dunst J, Winkelann W, Exner GU, Kotz R, et al. Localized Ewing tumor of bone: final results of the Cooperative Ewing Sarcoma Study CESS 86. J Clin Oncol 2001;19(6):1818-1829
-
Gu M, Antonescu CR, Guiter G, Huvos AG, Ladanyi M, Zakowski, MF: Cytokeratin immunoreactivity
in Ewing's sarcoma: Prevalence in 50 cases confirmed by molecular diagnostic studies. Am J Surg Path 24(3):410-416, 2000
|
|
|

